Regulatory
Data information and disclosure
Part 3
Author: Dr. Cheryl L. Rowe-Rendleman
Some exciting new changes were announced in 2023 that affect both drugs and Biologics. In the United States, “biologics” or “biological products” are subject to a different premarket pathway and differing intellectual property protections than products regulated as “drugs.”1 This blog series describes practices of the U.S. Food and Drug Administration (FDA), across key practices and processes for obtaining marketing approval for Biologic products.
Part 1: Obtaining Early Regulatory Advice-INTERACT Meetings
Part 2: Conducting Trials
Part 3: Data information and disclosure

In the last two blogs of this series, I discussed strategies behind holding meetings with the FDA to support the development of clinical trials for biologics.
In Part 1 initially early advice on biologic development using the INTERACT meeting was discussed. An INTERACT meeting is intended to facilitate IND-enabling efforts when the sponsor is facing a novel or challenging issue that might otherwise delay progress of the product toward entry into the clinic in the absence of this early FDA input.
In Part 2 the different types of clinical trials were discussed. In brief, the type of clinical studies to be conducted depends on the questions being asked, stage of development, risk factors, and standard of care. It is important to note that some of the most valuable clinical studies are Natural History studies that allow a sponsor to determine how a disease progresses in a specific population without intervention. This information is valuable because it can potentially indicate who will be the best candidate for an interventional trial with a biologic.
In this final section we consider the confidential and proprietary nature of informational meetings with the FDA and attempt to make an assessment of the types of information and disclosures that should be made to the FDA in order to get the best out of your early informational meetings with the Agency. Biologics are made from living cells. The type of data and disclosures made in your Interact and/or preIND meeting brief with the FDA must be carefully calibrated so that the FDA can make useful recommendations for development and clinical trials. The most sensitive data for a biologic is its manufacturing process.
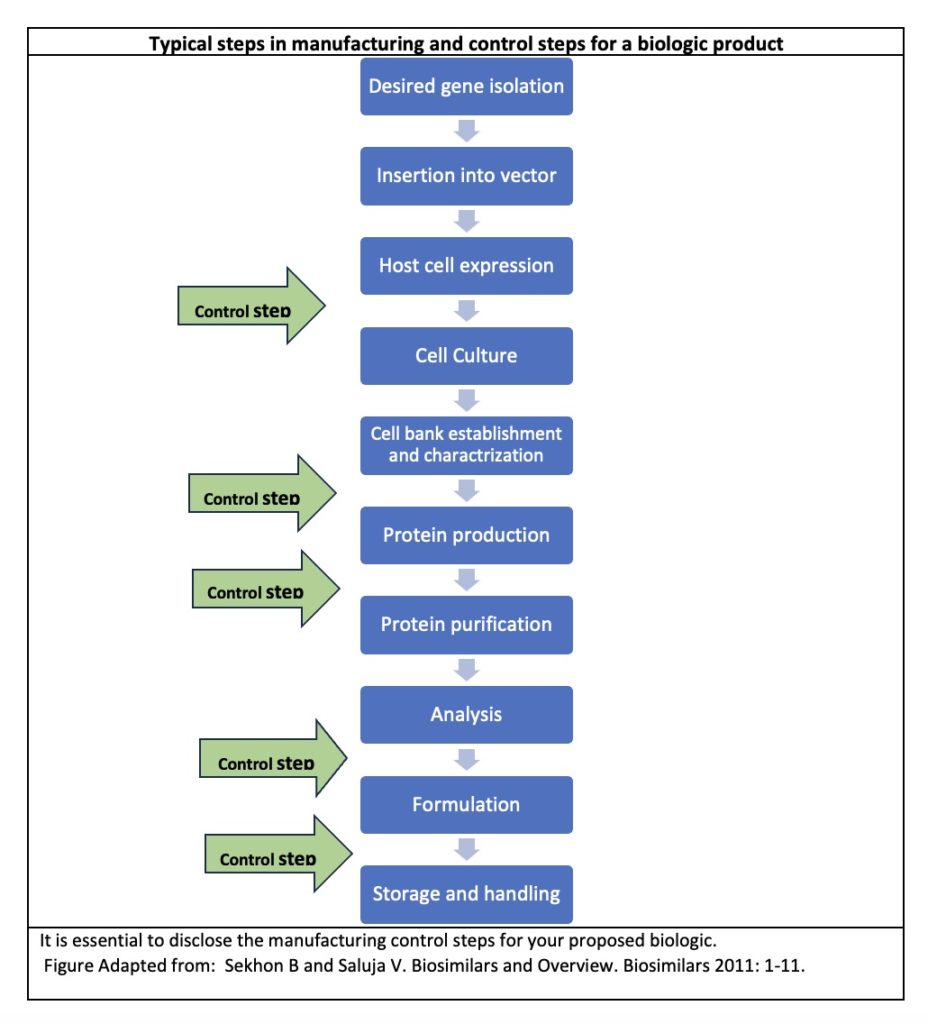
When disclosing manufacturing information about your biologic to the FDA, it is important to describe that your methods for derivation and isolation of the cells and manufacturing, control and purification steps for the product adhere to FDA standards. This is important because FDA enforces regulations necessary to prevent the introduction, transmission, or spread of communicable diseases from biologics.2
Manufacturers of certain biologics are subject to cGMP (current good manufacturing practice) regulations as well as to CGTP (current good tissue practice) regulations.3 FDA will remind sponsors to adhere to these practices during the INTERACT or preIND meetings. When the IND is reviewed, those who fail to follow these regulations make experience a hold on their proposed clinical study.
Even when a sponsor hires a contract manufacturing organization to produce their biologic, it is the sponsor’s responsibility to make sure that all cGMP and cGTP regulations are followed. The FDA advises that “before entering into a contract, agreement, or other arrangement with another establishment to perform any step in manufacture …you must ensure that the establishment complies with applicable cGTP requirements.”
In 2024 we can look forward to many more guidances for biologics from the FDA
| Guidance Documents* CBER is Planning to Issue in 2024 for Therapeutic Biologics4 |
| Human Gene Therapy Products Incorporating Human Genome Editing; Guidance for Industry |
| Considerations for the Development of Chimeric Antigen Receptor T Cell Products; Guidance for Industry |
| Frequently Asked Questions — Cell and Gene Therapy Products; Draft Guidance for Industry |
| Considerations for the Use of Human- and Animal- Derived Materials and Components in the Manufacture of Cell and Gene Therapy and Tissue-Engineered Medical Products; Draft Guidance for Industry |
| Safety Testing of Human Allogeneic Cells Expanded for Use in Cell-Based Medical Products; Draft Guidance for Industry |
| Recommendations for Determining Eligibility of Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps); Draft Guidance for Industry |
| Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products; Guidance for Industry |
| Potency Assurance for Cellular and Gene Therapy Products, Guidance for Industry |
| Recommendations to Reduce the Risk of Transmission of Mycobacterium tuberculosis by Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps), Guidance for Industry |
| Recommendations to Reduce the Risk of Transmission of Disease Agents Associated with Sepsis for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps), Guidance for Industry |
| In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word “should” in Agency guidances means that something is suggested or recommended, but not required. |
References
- Kingham R, Klasa G, and Carver KH . Key Regulatory Guidelines in the development of biologics in the United States and Europe. In Biological Drug Products: Development and Strategies. Editors Wei Wang and Manmohan Singh (2014). John Wiley & Sons, Publishers
- Federal Register / Vol. 69, No. 226 / Wednesday, November 24, 2004 / Rules and Regulations 04-25798.pdf (govinfo.gov)
- Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps); Guidance for Industry (fda.gov) . Accessed at https://www.fda.gov/ on February 2, 2024.
- Guidance Agenda: Guidance Documents CBER is Planning to Publish During Calendar Year 2024 (fda.gov). Accessed at https://www.fda.gov/ on February 2, 2024.
About the Author:
Omar Consulting Group, LLC

Dr. Cheryl Rowe-Rendleman serves as CEO and Managing Consultant at Omar Consulting Group, LLC. (Princeton NJ ǀ Durham, NC). Since 2006 Omar (ophthalmic management and research) specializes in regulatory and clinical consulting for drugs and devices in the US, Europe, and China. At Omar, Cheryl specializes in taking companies to the FDA and plays a leading role in developing successful paths to market and acquisition for emerging biotechnology.